

Abstract
Introduction
Hermansky-Pudlak syndrome (HPS) is an inherited platelet disorder characterized by bleeding diathesis, oculocutaneous albinism (OCA) and, sometimes, serious clinical complicationssuch as immunodeficiency, granulomatous colitis, and/or pulmonary fibrosis. Heterogeneous clinical symptoms and a large number of possible genetic culprits (10 HPS genes, >120 exons) complicate an unequivocal diagnosis of HPS. This study aimed to assess the clinical and platelet phenotype in ten patients with suspected HPS, and to identify the underlying genetic defects.
Methods
Ten patients from six families (F1 and F3 were Spanish, F2 was Turkish and F4, F5 and F6 were Portuguese) presenting with OCA (confirmed by skin biopsy) and bleeding diathesiswere included. Bleeding was evaluated by ISTH-BAT score. Phenotyping included, in patients with fresh blood samples available, platelet aggregation and ATP release, flow cytometry (FC), 14C-serotonin uptake and whole-mount electron microscopy (EM). Patients DNA was analyzed using two different targeted panels by high throughput sequencing (HTS). Sequence variants classification was performed according to ACMP recommendations.
Results
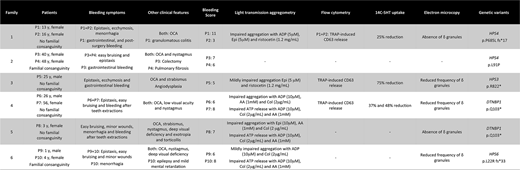
Patient characteristics are summarized in table 1. In F1, that had no history of consanguinity, there were two affected sisters. Patients 1 (P1) had several episodes of gastrointestinal bleeding (GI), which was attributed to granulomatous colitis. F2 is a consanguineous Turkish family, were P3 had severe rectal bleeding, requiring colectomy combined with ileostomy surgery. Pathological examination of the colon was reported as non-granulomatous colitis. Her older sister (P4) had exhibited dyspnea and shortness according to diffuse bilateral pulmonary fibrosis (BPF) diagnosis. In F3, P5 had been referred with acute GI bleeding secondary to angiodysplasia. In the non-consanguineous F4, HPS was first confirmed in P6, who showed blonde hair, nystagmus and low visual acuity; his older sister was diagnosed with HPS later, at the age of 56 years old (P7), because her OCA was masked using dark brown hair-coloring products. In P8, born from a non-consanguineous family (F5), HPS was suspected early in life, four months of age, upon recognition of OCA, nystagmus, deep visual deficiency and exotropia with compensatory torticollis. Lastly, in the consanguineous Portuguese family (F6), the two affected children (P9 and P10) had also showed a horizontal and torsional nystagmus and reduced visual activity. P10 also suffered from epilepsy and mild development delay. In phenotyping studies, the Spanish patients (P1, P2, P5) showed impaired platelet aggregation to mild agonists and reduced platelet dense granules by FC and EM. No platelet studies could be performed in F2. In Portuguese patients (F4, F5 and F6), the ATP release studies demonstrated a dense granule deficiency (Table 1). Molecular diagnosis was achieved, as a first-line approach, by means of HTS gene panels that revealed: a) F1 (P1 & P2) a homozygous deletion c.2054delC (p.P685L fs17*) in exon 13 of the HPS4, which had been previously reported in one Asian patient who showed BPF; b) F2 (P3 & P4): anovel missense homozygousvariant c.272T>C (p.L91P) in exon 4 of the HPS4. Remarkably, the phenotype of the two Turkish sisters was different, with one having had severe GI bleeding requiring colectomy, and the other had developed BPF. C) F3 (P5): a novel heterozygous variant c.2464C>T (p.R822*) in exon 13 of the HPS3 was detected; d) F4 (P6 & P7) and F5 (P8): here a nonsense variant c.307C>T (p.Q103*) was identified in exon 5 of the DTNBP1, which was previously reported in a Portuguese patient. E) F6 (P9 & P10): these patients carried a novel five base pair duplication in the single exon of HPS6, c.60_64dup (p.L22R fs*33).
Conclusions
This study reports 10 new HPS patients, which demonstrates the heterogeneous nature of this syndrome and the complex phenotype-genotype correlations. The novel HTStechnology has facilitated the molecular diagnosis of HPS in these patients. Among the underlying molecular pathology, we identified a novel p.L91P variant in HPS4 that is associated with a severe clinical phenotype.
Funding
Gerencia Regional de Salud (GRS 1647/A/17), Fundación Séneca (19873/GERM/15), Instituto de Salud Carlos III (ISCIII, PI17/01966, PI17/01311,CB15/00055), Grupo de trabajo SETH and Instituto de Investigación Biomédica de Salamanca (IBSAL, IBY17/00006).
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal